

ADAM 0.23.0 Released (+ Avocado and DECA Releases)
We are excited to announce the availability of the ADAM 0.23.0 release, along with releases of Avocado germline variant caller (release 0.1.0) and the DECA copy number variant caller (release 0.2.0). These releases contain an extensive number of feature additions, performance improvements, and bug patches, with over 375 issues closed and pull requests merged or closed since the last ADAM release.
Some of the highlights include:
- A validated, high-performance end-to-end alignment/variant calling pipeline using ADAM, Cannoli, and Avocado.
- Support for manipulating data using Spark SQL.
- R and Python APIs for ADAM, including the ability to get a working deployment
of ADAM simply by running
pip install bdgenomics.adam.
With this release, we have also moved our documentation to Read The Docs:
This documentation describes how to deploy our tools on a variety of platforms,
including a local cluster, cloud computing, and through the
Toil workflow manager. We already have
a pip installable Toil workflow for calling copy number variants with DECA,
which is packaged as part of the
bdgenomics.workflows library.
This release is the last release of ADAM that supports Spark 1.x and Scala 2.10. The upcoming release of ADAM will only support Spark 2.x and Scala 2.11. Avocado and DECA have already dropped support for Spark 1.x.
Over the upcoming few weeks, we are working on a release of Cannoli, as well as Toil workflows for running the ADAM/Avocado/Cannoli variant calling pipeline, and a preprint describing the pipeline in more depth. We also are working on a release of the Mango visualization tool, which uses ADAM as a backend for interactively visualizing large genomics datasets. Stay tuned for more info!
Variant Calling with Cannoli, ADAM, Avocado, and DECA
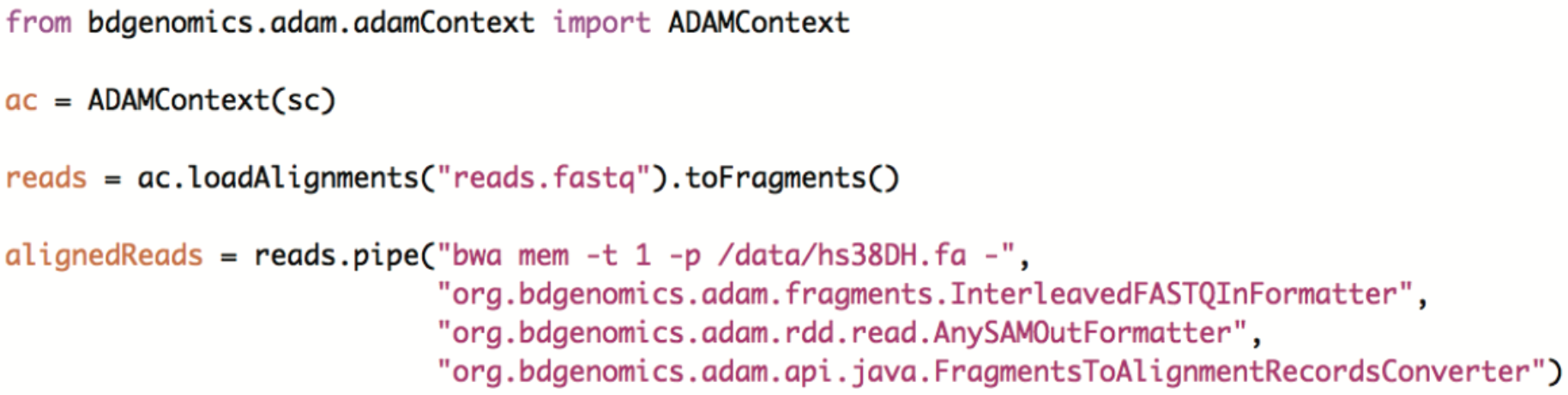
With the collection of tools we have released, you can run highly rapid and accurate variant calling entirely in Apache Spark. While we have introduced Avocado and DECA earlier in this post, we haven’t talked about Cannoli yet. Cannoli—-Italian for “a little pipe”—-uses ADAM’s pipe API to parallelize commonly used genomics tools. Currently, Cannoli supports aligning reads with Bowtie, Bowtie2, and BWA; calling variants with FreeBayes; and annotating variant effects with SnpEff. We are working on support for many more tools, as you can see in our issue tracker. Please let us know if you are interested in any specific tool—-or even better—-in helping us add support for a specific tool. ADAM’s pipe API makes it extremely easy to parallelize an existing single node genomic analysis tool, and most tools can be implemented on top of the pipe API in less than 10 lines of code. For example, here’s how you could launch BWA using ADAM’s Pipe API in Python:
By using Cannoli, we can accelerate alignment with BWA to take approximately 10—15 minutes when running on a 1,024 core cluster.
We can couple this rapid alignment pipeline with the fast preprocessing stages in ADAM and the variant calling stages in Avocado to call variants on a 60x coverage WGS dataset in approximately 45 minutes on a 1,024 core cluster. Avocado can be used to call variants on a single sample, or to jointly call variants using a gVCF-based workflow. When running on 1,024 cores, we were able to jointly genotype more than 10TB of gVCFs within approximately 6 hours. Avocado has >99% accuracy when genotyping SNPs, and >96% accuracy when genotyping INDELs. Detailed benchmarking results can be found in Chapter 8 of this thesis. Avocado is two times faster than the GATK4’s Spark-based implementation of the HaplotypeCaller, although it is worth pointing out that this is an unfair comparison, as the HaplotypeCaller performs local reassembly, while Avocado does not.
One interesting comparison is between the duplicate marking and BQSR tools in ADAM and in the GATK4. In both cases, ADAM’s implementation is faster than the GATK4’s equivalent implementation.


We have work-in-progress towards a Spark SQL-based implementation of duplicate marking, which will provide an additional >20% performance improvement. We hope to introduce this new duplicate marker in the 0.24.0 release of ADAM.
Manipulating Data using Spark SQL
Since Apache Spark 1.6, there has been a major push in the Spark project to rearchitect Spark around the Catalyst query optimizer and the Tungsten code execution engine. These two engines are hidden behind Spark SQL’s DataFrame and Dataset APIs, which provide a SQL-like interface for manipulating data using Spark. Unlike Spark’s Resilient Distributed Dataset (RDD) API, the DataFrame API allows the Catalyst query optimizer to examine the function that the user is running. Catalyst can then rewrite the query so that it runs in a more efficient manner, and can implement the query using the Tungsten engine with performance that approaches native performance. This can provide order-of-magnitude performance improvements for some queries, and it also provides users with uniform query performance across Scala, Java, SQL, Python, and R.
Although Spark SQL was introduced in 2015, we were not able to take advantage
of Spark SQL in ADAM until recently. While ADAM has always described genomics
data using a set of schemas, the library we used to represent these schemas
(Apache Avro) was not compatible with Spark SQL. To
resolve this, we updated our core GenomicRDD interfaces
to transparently convert between Spark’s RDD and DataFrame/Dataset APIs. We
describe the architecture we use for converting between these two representations
here.
With the Spark SQL query interfaces built into GenomicRDDs, you can begin
running SQL queries on genomic data in fewer than 5 lines of code:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 | |
While Spark SQL has specific optimizations for loading data from Apache Parquet files, ADAM can be used to run Spark SQL queries against data stored in most common genomics file formats, including SAM/BAM/CRAM, FASTQ, VCF/BCF, BED, GTF/GFF3, IntervalList, NarrowPeak, FASTA and more.
Using ADAM through Python and R
As mentioned above, one of the major advantages of Spark SQL is that it provides
uniform query performance across Scala, Java, Python, and R. While ADAM is
mostly written in Scala, we have maintained Java APIs for a long time. However,
we have previously been unable to support Python or R APIs. Adding support
for Spark SQL eliminated the major issues that prevented us from adding Python
and R APIs. This release of ADAM introduces the bdgenomics.adam packages for
Python and R. Our Python API can be installed using pip install
bdgenomics.adam, and our R API is available from
GitHub.
We hope to make our R API available through CRAN in the 0.24.0 release of ADAM;
we are blocked on an issue upstream in Apache Spark and are tracking progress on
this issue at ADAM-1851.
In addition to installing the bdgenomics.adam libraries, running pip install
bdgenomics.adam installs all of the ADAM command line tools:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 | |
Most of the major APIs in ADAM can be used through our Python and R bindings, with the exception of the region join API. We plan to enable the use of the region join API in Python and R in the 0.24.0 release of ADAM, along with other API compatibility improvements.
Changes since Previous Release
The full list of changes since version 0.22.0 is below.
Closed issues:
- Readthedocs build error #1854
- Add pip release to release scripts #1847
- Publish scaladoc script still attempts to build markdown docs #1845
- Allow variant annotations to be loaded into genotypes #1838
- Specify correct extensions for SAM/BAM output #1834
- Fix link anchors and other issues in readthedocs #1822
- Sphinx fulltoc is not included #1821
- Readme link to bigdatagenomics/lime 404s #1819
- Bump to Hadoop-BAM 7.9.1 #1817
- LoadVariants Header Format #1815
- Right and Left Outer Shuffle Region Join don’t match #1813
- Pipe command can fail with empty partitions #1807
- adam files with outdated formats throw FileNotFoundException #1804
- Move GenomicRDD.writeTextRDD outside of GenomicRDD #1803
- find-adam-assembly fails to recognize more than 1 jar #1801
- tests/testthat.R failed on git head #1799
- Run python and R tests conditionally in build #1795
- scala-lang should be a provided dependency #1789
- loadIndexedBam does an unnecessary union #1784
- Release bdgenomics.adam R package on CRAN #1783
- Issue with transformVariant // Adam to vcf #1782
- Add code of conduct #1779
- Reinstantiation of SQLContext in pyadam ADAMContext #1774
- Genotypes should only contain the core variant fields #1770
- Add SingleFASTQInFormatter #1768
- INDEL realigner can emit negative partition IDs #1763
- Request for a new release #1762
- INDEL realigner generates targets for reads with more than 1 INDEL #1753
- Fragment Issue #1752
- Variant Caller!!! #1751
- Spark Version!! #1750
- ReferenceRegion.subtract eliminating valid regions #1747
- New Shuffle Join Implementation – Left Outer + Group By Left #1745
- command failure after build success #1744
- Recalibrate_base_Qualities #1743
- Standardize regionFn for ShuffleJoin returned objects #1740
- Shuffle, Broadcast Joins with threshold #1739
- Adam on Spark 2.1 #1738
- Opening up permission on GenericGenomicRDD constructor #1735
- Consistency on ShuffleRegionJoin returns #1734
- vcf2adam support #1731
- Cloud-scale BWA MEM #1730
- Aligned Human Genome couldn’t convert to Adam #1729
- Mark Duplicates #1726
- Genomics Pipeline #1724
- .fastq Alignment #1723
- Is it correct Adam file #1720
- .fastQ to .adam #1718
- Unable to create .adam from .sam #1717
- Add adam- prefix to distribution module name #1716
- Python load methods don’t have ability to specify validation stringency #1715
- NPE when trying to map loadVariants over RDD #1713
- Add left normalization of INDELs as an RDD level primitive #1709
- Allow validation stringency to be set in AnySAMOutFormatter #1703
- InterleavedFastqInFormatter should sort by readInFragment #1702
- Allow silencing the # of reads in fragment warning in InterleavedFastqInFormatter #1701
- GenomicRDD.toXxx method names should be consistent #1699
- Exception thrown in VariantContextConverter.formatAllelicDepth despite SILENT validation stringency #1695
- Make GenomicRDD.toString more adam-shell friendly #1694
- Add adam-shell friendly VariantContextRDD.saveAsVcf method #1693
- change bdgenomics.adam package name for adam-python to bdg-adam #1691
- Conflict in bdg-formats dependency version due to org.hammerlab:genomic-loci #1688
- Convert and store variant quality field. #1682
- Region join shows non-determinism #1680
- Shuffle region join throws multimapped exception for unmapped reads #1679
- Push validation checks down to INFO/FORMAT fields #1676
- IndexOutOfBounds thrown when saving gVCF with no likelihoods #1673
- Generate docs from R API for distribution #1672
- Support loading a subset of VCF fields #1670
- Error with metadata: Multivalued flags are not supported for INFO lines #1669
- Include bdg.adam-0.23.0.tar.gz in distribution tarballs #1668
- Include bdgenomics.adam-0.23.0_SNAPSHOT-py2.7.egg in distribution tarball #1667
- Add SUPPORT.md file to complement CONTRIBUTING.md #1664
- Can’t merge BAM files containing the same sample #1663
- Incorrect README.md kmer.scala loadAliments method parameter name #1662
- Add performance benchmarks similar to Samtools CRAM benchmarking page #1661
- Transient bad GZIP header bug when loading BGZF FASTQ #1658
- bdgenomics.adam vs bdg.adam for R/Python APIs #1655
- Need adamR script #1649
- incorrect grep for assembly jars in bin/pyadam #1647
- VariantRDD union creates multiple records for the same SNP ID #1644
- S3 access documentation #1643
- Algorithms docs formatting #1639
- Building downstream apps docs reformatting #1638
- FastqInputFormat.FILE_SPLITTABLE in conf not getting passed properly #1635
- Add benchmarks to documentation #1634
- Intro docs contain outdated/incompatible code #1633
- Intro docs missing a number of active projects #1632
- Installation instructions for Homebrew missing from documentation #1631
- Architecture section is missing from docs #1630
- Seq
vs. Seq with javac #1625 - ProcessingStep missing from adam-codegen #1623
- Add ADAM recipe to bioconda #1618
- adam-submit cannot find assembly jar if installed as symlink #1616
- Expose transform/transmute in Java/Python/R #1615
- Expose VariantContextRDD in R/Python #1614
- Expose pipe API from Python/R #1611
- Serialization issue with TwoBitFile #1610
- Snapshot Distribution Does not include jar files #1607
- ManualRegionPartitioner is broken for ParallelFileMerger codepath #1602
- VariantRDD doesn’t save partition map #1601
- Scala copy method not supported in abstract classes such as AlignmentRecordRDD #1599
- Interleaved FASTQ recognizes only /1 suffix pattern #1589
- Use empty sequence dictionary when loading features #1588
- New Illumina FASTQ spec adds metadata to read name line #1585
- first run of ADAM #1582
- Add unit test coverage for BED12 parser and writer #1579
- Spark 1.x Scala 2.10 snapshot artifacts missing since 31 March 2017 #1578
- Unable to save GenomicRDDs after a join. #1576
- Add filterBySequenceDictionary to GenomicRDD #1575
- Unaligned Trait does nothing #1573
- Bump to bdg-formats 0.11.1 #1570
- PhredUtils conversion to log probabilities has insufficient resolution for PLs #1569
- Reference model import code is borked #1568
- SequenceDictionary vs Feature[RDD] of reference length features #1567
- giab-NA12878 truth_small_variants.vcf.gz header issues #1566
- VCF header read from stream ignored in VCFOutFormatter #1564
- VCF genotype Number=A attribute throws ArrayIndexOutOfBoundsException #1562
- Save compressed single file VCF via HadoopBAM #1554
- bucketing strategy #1553
- Is parquet using delta encoding for positions? #1552
- Export to VCF does not include symbolic non-ref if site has a called alt #1551
- Refactor filterByOverlappingRegions not to require a List #1549
- Move docs to Sphinx/pure Markdown #1548
- java.lang.IncompatibleClassChangeError: Implementing class #1544
- Support locus predicate in
TransformAlignments#1539 - Visibility from Java, jrdd has private access in AvroGenomicRDD #1538
- Rename o.b.adam.apis.java package to o.b.adam.api.java #1537
- VCF header genotype reserved key FT cardinality clobbered by htsjdk #1535
- Compute a SequenceDictionary from a *.genome file #1534
- Queryname sorted check should check for queryname grouped as well #1530
- Bump to bdg-formats 0.11.0 #1520
- Move to Spark 2.2, Parquet 1.8.2 #1517
- Minor refactor for TreeRegionJoin for consistency #1514
- Allow +Inf and -Inf Float values when reading VCF #1512
- SparkFiles temp directory path should be accessible as a variable #1510
- SparkFiles.get expects just the filename #1509
- Split apart #1324 #1507
- Where can I find “Phred-scaled quality score” (QUAL)? #1506
- Alignment Record sort is not consistent with samtools #1504
- Sequence dictionary records in TwoBitFile are not stable #1502
- Move coverage counter over to Dataset API #1501
- Allow users to set the minimum partition count across all load methods #1500
- Enable reuse of broadcast object across broadcast region joins #1499
- Take union across genomic RDDs #1497
- Adam files created by vcf2adam is not recognizable #1496
- Scalatest log output disappears with Maven 3.5.0 #1495
- ArrayOutOfBoundsException in vcf2adam (spark2_2.11-0.22.0) on UK10K VCFs (VCFv4.1) #1494
- ReferenceRegion overlaps and covers returns false if overlap is 1 #1492
- Provide asSingleFile parameter for saveAsFastq and related #1490
- Min Phred score gets bumped by 33 twice in BQSR #1488
- Should throw error when BAM header load fails #1486
- Default value for reads.toCoverage(collapse) should be false #1483
- Refactor ADAMContext loadXxx methods for consistency #1481
- loadGenotypes three time #1480
- Fall back to sequential concat when HDFS concat fails #1478
- VCF line with
.ALT gets dropped #1476 - ADAM works on Cloudera but does NOT work on MAPR #1475
- Clean up ReferenceRegion.scala #1474
- Allow joins on regions that are within a threshold (instead of requiring overlap) #1473
- FeatureRDD.toCoverage throws NullPointerException when there is no coverage information #1471
- Add quality score binner #1462
- Splittable compression and FASTQ #1457
- Don’t convert .{different-type}.adam in loadAlignments and loadFragments #1456
- New primitives for adam-core #1454
- Port over code for populating SequenceDictionaries from .dict files #1449
- Ignore failed push to Coveralls during CI builds #1444
- No asSingleFile parameter for saveAsFasta in NucleotideContigFragmentRDD #1438
- shufflejoin and ArrayIndexOutOfBoundsException #1436
- Document using ADAM snapshot #1432
- Improve metrics coverage across ADAMContext load methods #1428
- loadReferenceFile missing from Java API #1421
- loadCoverage missing from Java API #1420
- Question: How to get paired-end alignemntRecord like RDD[AlignmentRecord, AlignmentRecordRDD]? #1419
- Clean up possibly unused methods in Projection #1417
- Problem loading SNPeff annotated VCF #1390
- RecordGroupDictionary should support
isEmpty#1380 - Get rid of mutable collection transformations in ShuffleRegionJoin #1379
- Add tab5/6 as native output format for AlignmentRecordRDD #1377
- ValidationStringency in MDTagging should apply to reads on unknown references #1365
- Assembly final name doesn’t include spark2 for Spark 2.x builds #1361
- Merge reads2fragments and fragments2reads into a single CLI #1359
- Investigate failures to load ExAC.0.3.GRCh38.vcf variants #1351
- adam-shell does not allow additional jars via Spark jars argument #1349
- Loading GZipped VCF returns an empty RDD #1333
- Bump Spark 2 build to Spark 2.1.0 #1330
- Rename Transform command TransformAlignments or similar #1328
- Replace ADAM2Vcf and Vcf2ADAM commands with TransformGenotypes and TransformVariants #1327
- FeatureRDD instantiation tries to cache the RDD #1321
- Repository for Pipe API wrappers for bioinformatics tools #1314
- Trying to get Spark pipeline working with slightly out of date code. #1313
- Support for gVCF merging and genotyping (e.g. CombineGVCFs and GenotypeGVCFs) #1312
- Support for read alignment and variant calling in Adam? (e.g. BWA + Freebayes) #1311
- Don’t include log4j.properties in published JAR #1300
- Removing ProgramRecords info when saving data to sam/bam? #1257
- ADAM on Slurm/LSF #1229
- Maintaining sorted/partitioned knowledge #1216
- Evaluate bdg-convert external conversion library proposal #1197
- Port AMPCamp Tutorial over #1174
- Top level WrappedRDD or similar abstraction #1173
- GFF3 formatted features written as single file must include gff-version pragma #1169
- Can probably eliminate sort in RealignIndels #1137
- Load SV type info field – need for allele uniquness #1134
- BroadcastRegionJoin is not a broadcast join #1110
- AlignmentRecordRDD does not extend GenomicRDD per javac #1092
- Add generic ReferenceRegion pushdown for parquet files #1047
- Use of dataset api in ADAM #1018
- Difference running markdups with and without projection #1014
- ADAM to BAM conversion fails using relative path #1012
- Refactor SequenceDictionary to use Contig instead of SequenceRecord #997
- NoSuchMethodError due to kryo minor-version mismatch #955
- Autogen field names in projection package #941
- Future of schemas in bdg-formats #925
- genotypeType for genotypes with multiple OtherAlt alleles? #897
- How to filter genotype RDD with FeatureRDD #890
- How to convert genotype DataFrame to VariantContext DataFrame / RDD #886
- R language package for Adam #882
- How to count genotypes with a 10 node Spark/Adam cluster faster than with BCFTools on a single machine? #879
- Ensure Java API is up-to-date with Scala API #855
- BroadcastRegionJoin fails with unmapped reads #821
- Resolve Fragment vs. SingleReadBucket #789
- Updating/Publishing the docs/ directory #774
- Next on empty iterator in BroadcastRegionJoin #661
- Cleanup code smell in sort work balancing code #635
- Provide low-impact alternative to
transform -repartitionfor reducing partition size #594 - Create an ADAM Python API #538
- Migrate serialization libraries out of ADAM core #448
- Create standardized, interpretable exceptions for error reporting #420
- Build info/version info inside ADAM-generated files #188
Merged and closed pull requests:
- [ADAM-1854] Add requirements.txt file for RTD. #1856 (fnothaft)
- [ADAM-1783] Resolve check issues that block pushing to CRAN. #1849 (fnothaft)
- [ADAM-1847] Update ADAM scripts to support self-contained pip install. #1848 (fnothaft)
- [ADAM-1845] Only build and publish scaladocs in publish-scaladoc.sh. #1846 (heuermh)
- [ADAM-1843] Install sources before calling scala:doc in publish scaladoc #1844 (fnothaft)
- Remove python and R profiles from release script #1842 (heuermh)
- [ADAM-1817] Bump to Hadoop-BAM 7.9.1. #1841 (fnothaft)
- [ADAM-1838] Make populating variant.annotation field in Genotype configurable #1839 (fnothaft)
- [ADAM-1834] Add proper extensions for SAM/BAM/CRAM output formats. #1835 (fnothaft)
- [ADAM-1822] Misc docs cleanup #1827 (fnothaft)
- Added missing init.py for fulltoc. #1824 (fnothaft)
- [ADAM-1821] Add missing fulltoc for Sphinx documentation. #1823 (fnothaft)
- Fix link to documentation #1820 (nzachow)
- [ADAM-1634] Add algorithm benchmarks to documentation. #1818 (fnothaft)
- [ADAM-1813] Delegate right outer shuffle region join to left OSRJ implementation. #1814 (fnothaft)
- [ADAM-1807] Check for empty partition when running a piped command. #1812 (fnothaft)
- [ADAM-1803] Refactor GenomicRDD.writeTextRdd to util.TextRddWriter. #1809 (heuermh)
- Added Filter error when file loaded does not match schema #1805 (akmorrow13)
- changed num_jars count #1802 (akmorrow13)
- [ADAM-1795] Map -DskipTests=true to exec.skip for Python and R tests. #1800 (heuermh)
- [ADAM-1672] Use working directory for R devtools::document(). #1798 (heuermh)
- [ADAM-1789] Move scala-lang to provided scope. #1790 (fnothaft)
- [ADAM-1784] loadIndexedBam should pass the raw globbed path to Hadoop-BAM #1785 (fnothaft)
- [ADAM-1664] Add SUPPORT.md file to complement CONTRIBUTING.md. #1781 (heuermh)
- [ADAM-1779] Adding code of contact adapted from the Contributor Convenant, version 1.4. #1780 (heuermh)
- [ADAM-1661] Add file storage benchmarks. #1772 (fnothaft)
- [ADAM-1770] Genotype should only store core variant fields. #1771 (fnothaft)
- [ADAM-1768] Add InFormatter for unpaired FASTQ. #1769 (fnothaft)
- [ADAM-1643] Add S3 access documentation. #1767 (fnothaft)
- [ADAM-1763] Apply absolute value to destination partition in ModPartitioner #1766 (fnothaft)
- Add R and Python into distribution artifacts #1765 (fnothaft)
- [ADAM-1655] Move R package to bdgenomics.adam. #1764 (fnothaft)
- [ADAM-1753] Only emit realignment targets for reads containing a single INDEL #1756 (fnothaft)
- [ADAM-1715] Support validation stringency in Python/R. #1755 (fnothaft)
- [ADAM-1680] Eliminate non-determinism in the ShuffleRegionJoin. #1754 (fnothaft)
- update to _replaceRdd with tests #1749 (akmorrow13)
- [ADAM-1747] Fixed subtract bug and tests #1748 (devin-petersohn)
- [ADAM-1745] Adding LeftOuterShuffleRegionJoinAndGroupByLeft and tests #1746 (devin-petersohn)
- Enabled thresholding for joins and standardized regionFn #1741 (devin-petersohn)
- Making join return types consistent #1737 (devin-petersohn)
- Opening up permissions on GenericGenomicRDD #1736 (devin-petersohn)
- [ADAM-1716] Add adam- prefix to distribution module name. #1733 (heuermh)
- [ADAM-1695] Check for illegal genotype index after splitting multi-allelic variants. #1725 (heuermh)
- [ADAM-1517] Bump Parquet version in a manner compatible with Spark 2.2.x #1722 (fnothaft)
- [ADAM-1512] Support VCFs with +Inf/-Inf float values. #1721 (fnothaft)
- [ADAM-1709] Add ability to left normalize reads containing INDELs. #1711 (fnothaft)
- [ADAM-1691] Move bdgenomics.adam to use a namespace package. #1706 (fnothaft)
- moved bdgenomics.adam package to bdgenomics-adam #1705 (akmorrow13)
- Misc cleanup needed for bigdatagenomics/cannoli#65 #1704 (fnothaft)
- [ADAM-1699] Make GenomicRDD.toXxx method names consistent. #1700 (heuermh)
- [ADAM-1694] Add short readable descriptions for toString in subclasses of GenomicRDD. #1698 (heuermh)
- [ADAM-1693] Add adam-shell friendly VariantContextRDD.saveAsVcf method. #1696 (heuermh)
- [ADAM-1688] Add bdg-formats exclusion to org.hammerlab:genomic-loci dependency. #1690 (heuermh)
- [ADAM-1679] Unmapped items should not get caught in requirement when sorting #1687 (fnothaft)
- [ADAM-1566] Merge VCF header lines with VCFHeaderLineCount.INTEGER correctly. #1685 (heuermh)
- [ADAM-1682] Add variant quality field. #1684 (fnothaft)
- Remove adam- prefix from module directory names. #1681 (heuermh)
- Update to hadoop-bam 7.9.0 and htsjdk 2.11.0. #1678 (heuermh)
- [ADAM-1676] Add more finely grained validation for INFO/FORMAT fields. #1677 (fnothaft)
- Python API fixes for AlignmentRecordRDD #1675 (akmorrow13)
- [ADAM-1673] Don’t set PL to empty when no PL is attached to a gVCF record #1674 (fnothaft)
- [ADAM-1670] Add ability to selectively project VCF fields. #1671 (fnothaft)
- [ADAM-1663] Enable read groups with repeated names when unioning. #1665 (fnothaft)
- Maint 2.11 0.18.0 #1659 (Douglas-H)
- [ADAM-1630] Overhauled docs introduction and added architecture section. #1653 (fnothaft)
- Add adamR script #1651 (fnothaft)
- [ADAM-1647] Fix bad JAR discovery grep in bin/pyadam. #1648 (fnothaft)
- [ADAM-1548] Generate reStructuredText from pandoc markdown. #1646 (fnothaft)
- Algorithms docs formatting #1645 (gunjanbaid)
- Cleaned up docs. #1642 (gunjanbaid)
- Making example code compatible with current ADAM build #1641 (devin-petersohn)
- Cleaning up formatting and spacing of docs. #1640 (devin-petersohn)
- added ExtractRegions #1637 (antonkulaga)
- [ADAM-1635] Eliminate passing FASTQ splittable status via config. #1636 (fnothaft)
- [ADAM-1614] Add VariantContextRDD to R and Python APIs. #1628 (fnothaft)
- [ADAM-1615] Add transform and transmute APIs to Java, R, and Python #1627 (fnothaft)
- [ADAM-1625] Use explicit types for header lines #1626 (heuermh)
- [ADAM-1623] Add ProcessingStep to adam-codegen. #1624 (heuermh)
- [ADAM-1607] Update distribution assembly task to attach assembly überjar #1622 (fnothaft)
- [ADAM-1490] Add asSingleFile to saveAsFastq and related. #1621 (heuermh)
- Update load method docs in Python and R. #1619 (heuermh)
- [ADAM-1616] Resolve installation directory if scripts are symlinks. #1617 (heuermh)
- [ADAM-1611] Extend pipe APIs to Java, Python, and R. #1613 (fnothaft)
- [ADAM-1610] Mark non-serializable field in TwoBitFile as transient. #1612 (fnothaft)
- [ADAM-1554] Support saving BGZF VCF output. #1608 (fnothaft)
- Adding examples of how to use joins in the real world #1605 (devin-petersohn)
- [ADAM-1599] Add explicit functions for updating GenomicRDD metadata. #1600 (fnothaft)
- [ADAM-1576] Allow translation between two different GenomicRDD types. #1598 (fnothaft)
- [ADAM-1444] Ignore failed push to Coveralls. #1595 (fnothaft)
- Testing, testing, 1… 2… 3… #1592 (fnothaft)
- [ADAM-1417] Removed unused Projection.apply method, add test for Filter. #1591 (fnothaft)
- [ADAM-1579] Add unit test coverage for BED12 format. #1587 (fnothaft)
- [ADAM-1585] Support additional Illumina FASTQ metadata. #1586 (fnothaft)
- [ADAM-1438] Add ability to save FASTA back as a single file. #1581 (fnothaft)
- Bump bdg-formats correctly to 0.11.1, not SNAPSHOT. #1577 (fnothaft)
- [ADAM-1573] Remove unused Unaligned trait. #1574 (fnothaft)
- Slurm deployment readme #1571 (jpdna)
- [ADAM-1564] Read VCF header from stream in VCFOutFormatter. #1565 (heuermh)
- [ADAM-1562] Index off by one for VCF genotype Number=A attributes. #1563 (heuermh)
- [ADAM-1533] Set Theory #1561 (devin-petersohn)
- Freebayes FORMAT=<ID=AO,Number=A attribute throws ArrayIndexOutOfBoundsException #1560 (heuermh)
- [ADAM-1551] Emit non-reference model genotype at called sites. #1559 (fnothaft)
- [ADAM-1449] Add loadSequenceDictionary to ADAM context. #1557 (heuermh)
- [ADAM-1537] Rename o.b.adam.apis.java package to o.b.adam.api.java #1556 (heuermh)
- [ADAM-1549] Make regions provided to filterByOverlappingRegions an Iterable. #1550 (fnothaft)
- [ADAM-941] Automatically generate projection enums. #1547 (fnothaft)
- [ADAM-1361] Fix misnamed ADAM überjar. #1546 (fnothaft)
- [ADAM-1257] Add program record support for alignment/fragment files. #1545 (fnothaft)
- [ADAM-1359] Merge
reads2fragmentsandfragments2readsintotransformFragments#1543 (fnothaft) - Fix minor format mistakes (and typo) in docs #1542 (kkaneda)
- Add a simple unit test to SingleFastqInputFormat #1541 (kkaneda)
- Support locus predicate in Transform #1540 (fnothaft)
- [ADAM-1421] Add java API for
loadReferenceFile. #1536 (fnothaft) - Refactor Vcf2ADAM and ADAM2Vcf into TransformGenotypes and TransformVariants #1532 (heuermh)
- [ADAM-1530] Support loading GO:query (S/CR/B)AMs as fragments. #1531 (fnothaft)
- [ADAM-1169] Write GFF header line pragma in single file mode. #1529 (fnothaft)
- [ADAM-1501] Compute coverage using Dataset API. #1528 (fnothaft)
- [ADAM-1497] Add union to GenomicRDD. #1526 (fnothaft)
- [ADAM-1486] Respect validation stringency if BAM header load fails. #1525 (fnothaft)
- [ADAM-1499] Enable reuse of broadcasted objects in region join. #1524 (fnothaft)
- [ADAM-1520] Bump to bdg-formats 0.11.0. #1523 (fnothaft)
- Adding fragment InFormatter for Bowtie tab5 format #1522 (heuermh)
- [ADAM-1328] Rename
TransformtoTransformAlignments. #1521 (fnothaft) - [ADAM-1517] Move to Parquet 1.8.2 in preparation for moving to Spark 2.2.0 #1518 (fnothaft)
- Fixed minor typos in README. #1516 (gunjanbaid)
- Making TreeRegionJoin consistent with ShuffleRegionJoin #1515 (devin-petersohn)
- Resolve #1508, #1509 for Pipe API #1511 (fnothaft)
- [ADAM-1502] Preserve contig ordering in TwoBitFile sequence dictionary. #1508 (fnothaft)
- [ADAM-1483] Remove collapse parameter from AlignmentRecordRDD.toCoverage #1493 (fnothaft)
- [ADAM-1377] Adding fragment InFormatter for Bowtie tab6 format #1491 (heuermh)
- [ADAM-1488] Only increment BQSR min quality by 33 once. #1489 (fnothaft)
- [ADAM-1481] Refactor ADAMContext loadXxx methods for consistency #1487 (heuermh)
- Add quality score binner #1485 (fnothaft)
- Clean up ReferenceRegion.scala and add thresholded overlap and covers #1484 (devin-petersohn)
- [ADAM-1456] Remove .{type}.adam file extension conversions in type-guessing methods. #1482 (heuermh)
- [ADAM-1480] Add switch to disable the fast concat method. #1479 (fnothaft)
- [ADAM-1476] Treat
.ALT allele as symbolic non-ref. #1477 (fnothaft) - Adding require for Coverage Conversion and related tests #1472 (devin-petersohn)
- Add cache argument to loadFeatures, additional Feature timers #1427 (heuermh)
- [ADAM-882] R API #1397 (fnothaft)
- [ADAM-1018] Add support for Spark SQL Datasets. #1391 (fnothaft)
- WIP Python API #1387 (fnothaft)
- [ADAM-1365] Apply validation stringency to reads on missing contigs when MD tagging #1366 (fnothaft)
- Update dependency and plugin versions #1360 (heuermh)
- [ADAM-1330] Move to Spark 2.1.0. #1332 (fnothaft)
- Efficient Joins and (re)Partitioning #1324 (devin-petersohn)